Research Overview
Molecular-scale structural and electronic interactions are responsible for the unique energy transfer properties found in biological light-harvesting systems. Captured energy from sunlight is transferred and harvested at remarkable efficiencies approaching unity in certain cases. This is due to the specific molecular organization of chromophores scaffolded by protein assemblies, yielding efficient short- and long-range energy transport. We probe these short- and long-range interactions of molecular assemblies using electronic structure theory and multi-scale modeling in order to learn from nature and apply these organizational concepts to synthetic energy transport systems.
Molecular Aggregate Structure
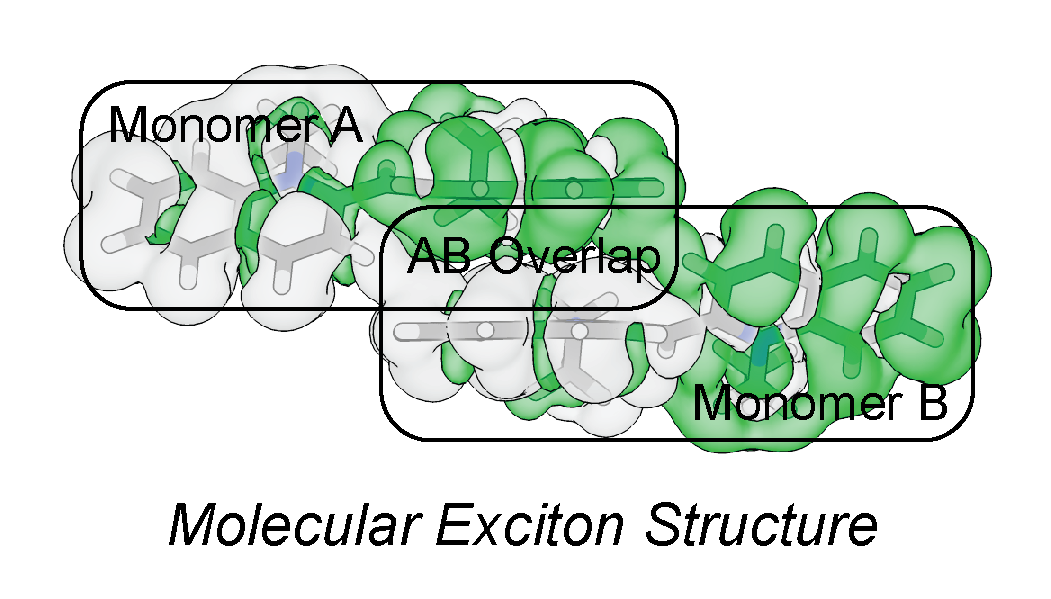
When chromophores aggregate, the electronic structure of an excited state can become delocalized over two or more molecular units creating an exciton. The relative orientation and magnitude of the molecule’s electronic transition dipoles will dictate the spectral and energy transfer properties of the exciton. By modifying the molecular aggregate structure, unique electronic and energy transfer properties can be designed. We aim to develop a computational toolset to calculate electronic interactions of molecular aggregates, and to predict their excitonic functionality.
Organization of Chromophore Assemblies
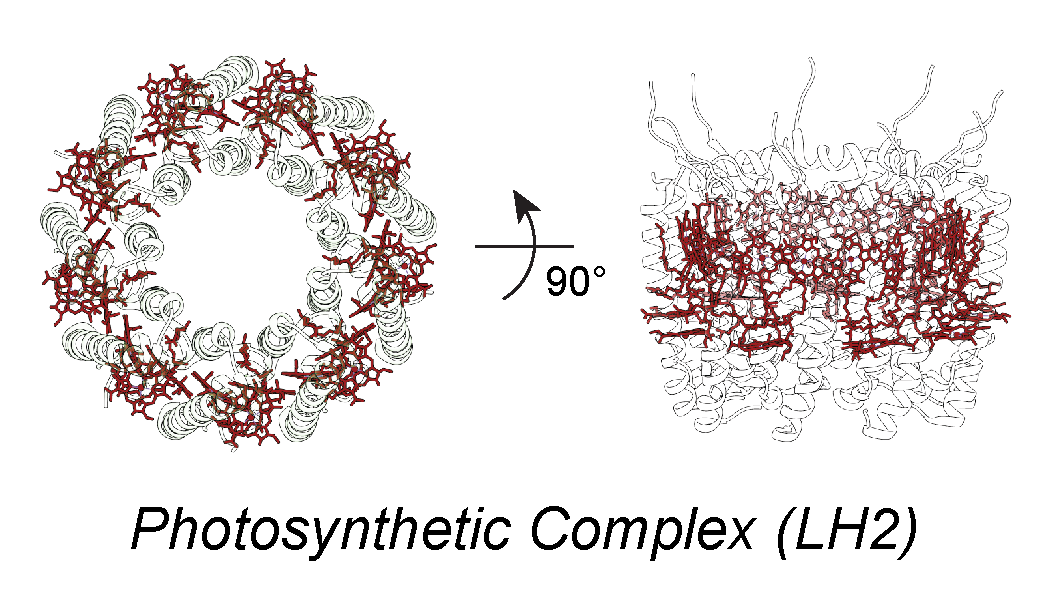
Biomolecular light-harvesting complexes in nature utilize a combination of short- and long-range energy transfer mechanisms to efficiently harvest sunlight and to avoid energy dissipation. In addition, different species of chromophores are present within the same complexes, yielding varying spectral properties in order to direct energy transfer via a downhill energetics funnel. With the inclusion of the relative orientation of molecules, the possible parameter space for designing biosynthetic energy transport systems is essentially intractable. We aim to reduce the parameter space by applying a systems-level optimization of chromophore organization to determine the relative importance of each parameter.
Electronic and Molecular Modeling
Using modern computational chemistry methods, including electronic structure theory and molecular dynamics, the unique properties of biomolecular systems can be probed. In addition, the more recent application of GPU hardware to biomolecular simulation has expanded the size and complexity of systems that can be modeled. We utilize existing computational chemistry software as well as develop our own modeling tools using modern scripting languages such as Python.
